How Bacteria Outsmart Our Best Drugs?
[Communications Biology | (2024) 7:1051]
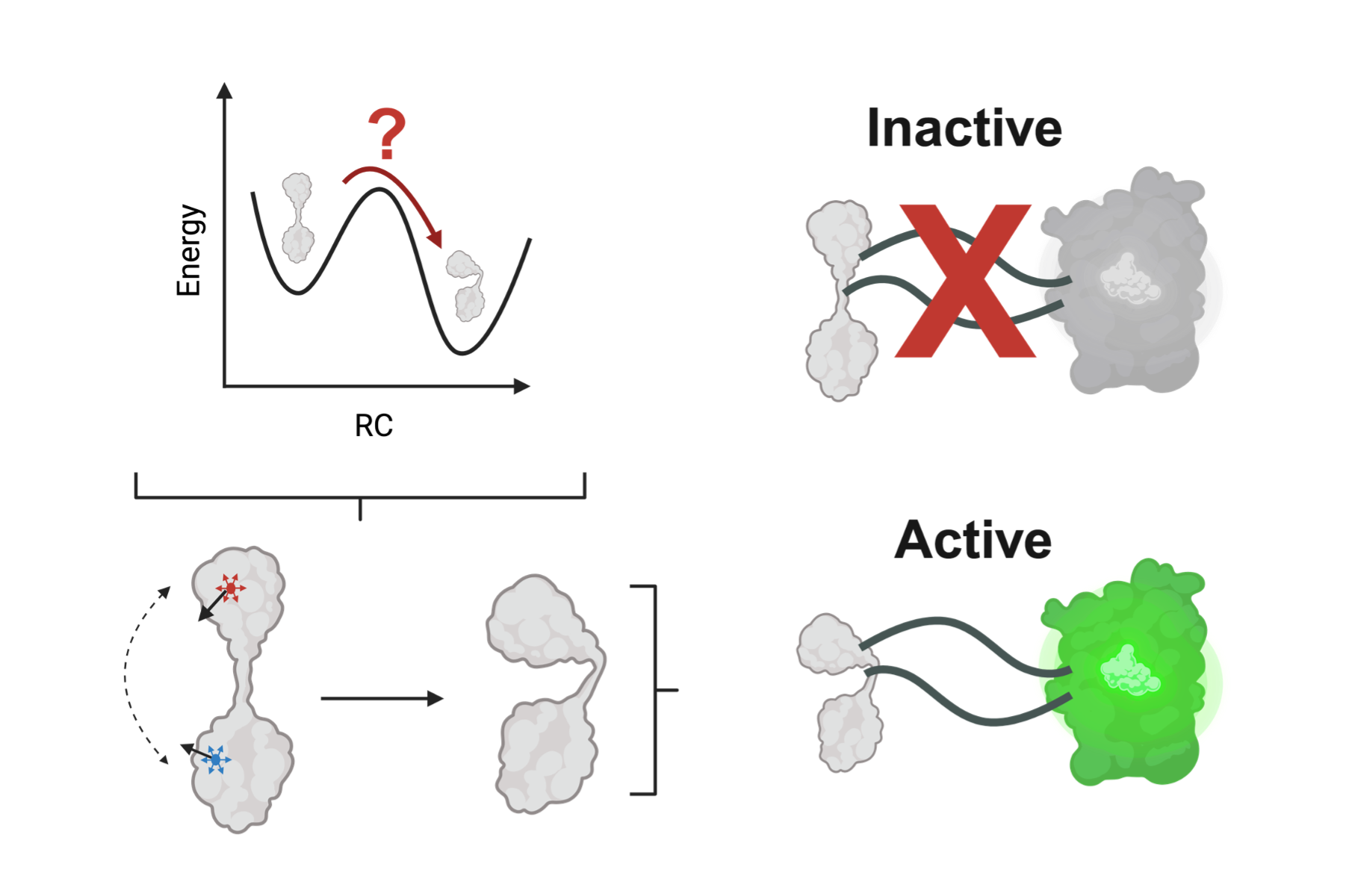
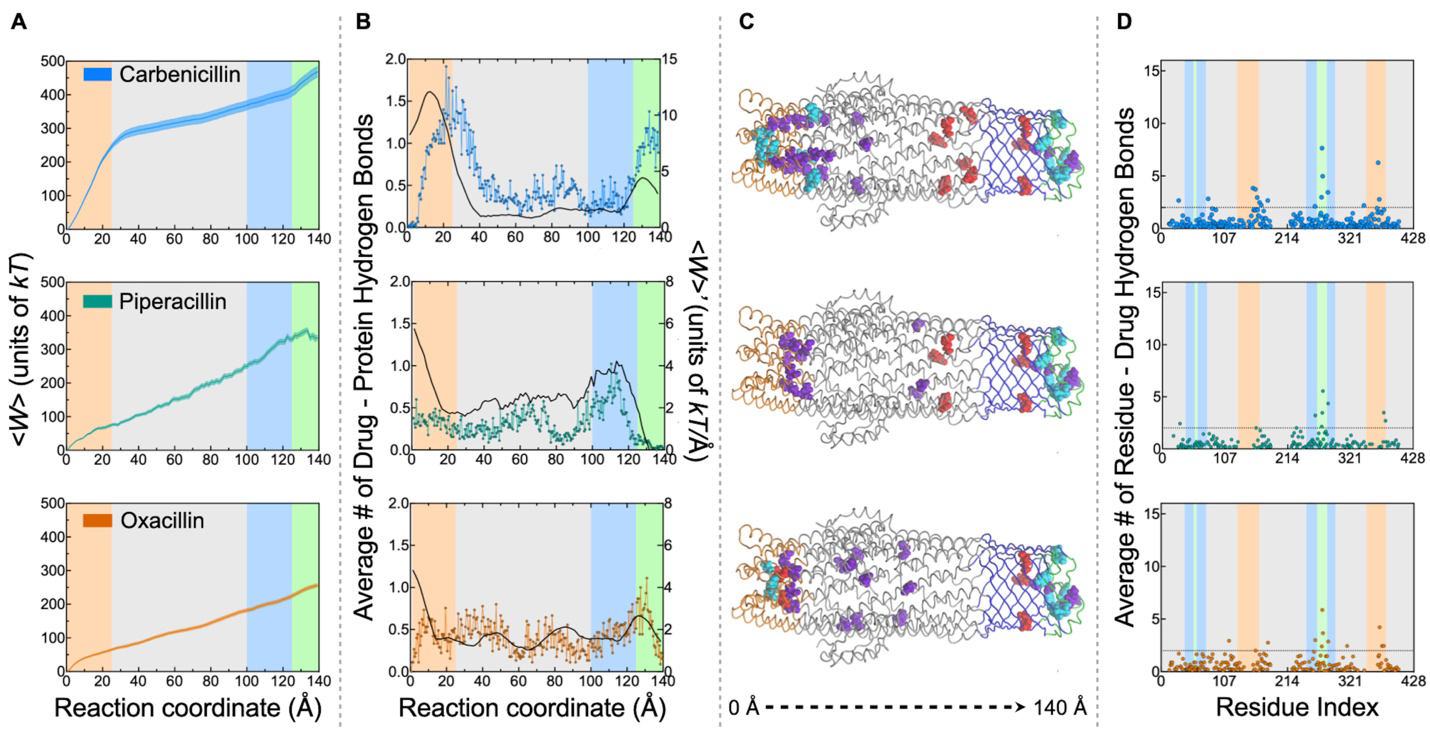
Structural shifts in a tiny channel protein help microbes pump out life saving antibiotics. Deep inside bacterial membranes, TolC acts as a molecular escape hatch, helping Gram negative bacteria expel compounds including β lactam antibiotics.
Read More